For RootsTech Connect this past week, we released the tool HAPI, which reconstructs DNA for one parent using data from three or more children and their other parent. Two videos from RootsTech describe ancestor DNA reconstruction in general and the HAPI tool in particular.
This post briefly reviews the video about HAPI and then goes into some technical details about it, including a few issues and questions that have come up from users in the past few days.
How much DNA can HAPI reconstruct and how accurately?
Parents transmit half of their DNA to each child, but it is a random half. One implication of this is that two siblings will have inherited different portions of their parent’s DNA, and those portions can be joined together to form more than 50% of the parent’s DNA. In fact, on average a parent transmits a fraction of 1 – ½C of their DNA to C children. But it’s worth noting that this is just on average: sometimes the parent transmits more, sometimes less (see below).
Parents transmit the same chromosome to all children at some locations.
One thing to keep in mind is that a parent transmits one DNA base pair (one “piece” of DNA) at every location to each child. So reconstructing in the way HAPI does will always get at least one copy of the parent’s two chromosome at every position (ignoring occasional errors or missing SNPs in the raw data for the children or other parent). But, as shown to the right, sometimes only one chromosome gets transmitted to all C children—here a pink colored chromosome. Therefore HAPI can only reconstruct one chromosome at some locations, and there are some technical concerns with this, as described in the next section.
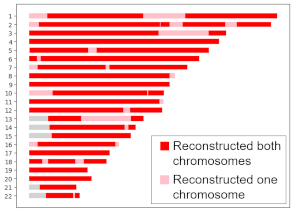
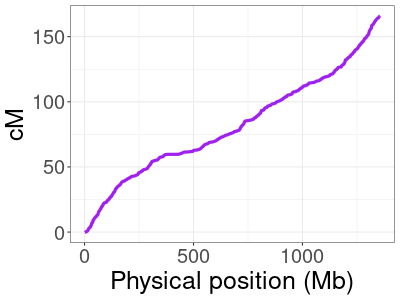
Locations where HAPI reconstructed my grandma on both or one copy.
My grandma died before genetic testing was widely available, but my dad, two aunts, one uncle, and my grandfather all consented to have their DNA tested. Using their raw data, HAPI reconstructed my grandma’s DNA, as shown on the right. For most locations, depicted in red, HAPI recovered data for both her chromosomes, but for about 10% of her DNA, HAPI recovered only one chromosome copy, as shown in pink. Overall, HAPI reconstructed 94.4% of her DNA, which is very close the average we would expect for C = 4 children, which is 93.75%, but that’s not so important—it could have been less or more.
To illustrate the variability of the amount of DNA reconstructed, we used DNA from 114 families from the San Antonio Mexican American Family Studies data (SAMAFS, analyzed here and other places) to reconstruct the father or the mother in each family, each parent in turn. The amount of reconstructed DNA in these parents varies widely, depending in part on the number of children in the family. The 114 families contain between 3 to 12 children (average 4.6), and the left plot shows the percent reconstructed in all 228 reconstructed parents (2 × 114). Focusing in on the two extremes, the top right plot gives the percent reconstructed from families with exactly 3 children, and the bottom right plot shows those with ≥ 8 children. With three children, we expect to recover 87.5% of the parent’s DNA, but in some families, we recover only 79% and in others we recover almost 92%! For large families, HAPI reconstructs nearly all of the parent’s DNA.
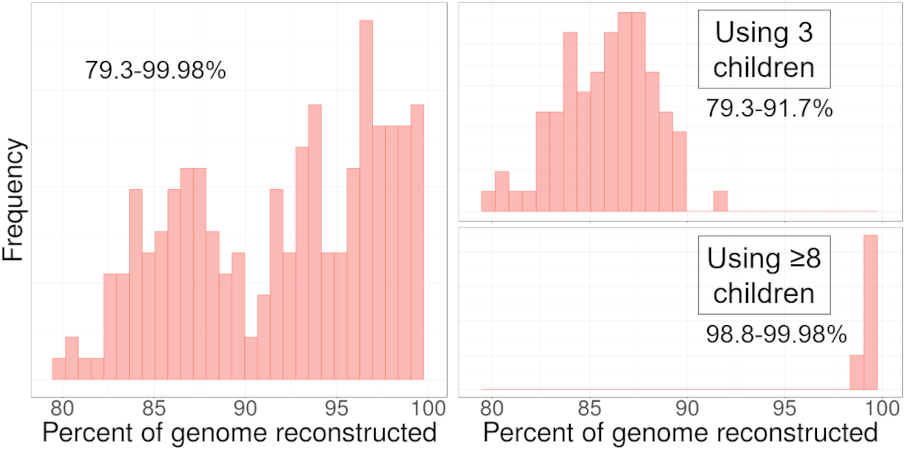
Histogram depicting the percentage of the parent’s DNA reconstructed in the SAMAFS data. Left: all families.
How accurate is the reconstructed DNA? The 114 families from the SAMAFS data actually include data from both parents, which we removed temporarily (both the father and mother in turn) to test HAPI. After running HAPI, we then compared the reconstructed DNA to the real data. The 228 reconstructed parents together have roughly 114 million SNPs, and, of those, only 36,338 differed from the truth, meaning that the reconstructed data was > 99% correct! A caveat to this is that the quality control performed on the SAMAFS data included many checks that aren’t possible to do with data from these companies, so the input data may be much more reliable than the raw data that company’s provide.
What does HAPI do in regions with only one reconstructed chromosome?
As noted above, except for cases with very large numbers of children, some portion of the parent’s DNA won’t have been transmitted and therefore there will be some parts of the genome where HAPI only recovers one of the two chromosomes. To make the printed kit readable by sites that allow uploads, HAPI currently makes two copies of the one reconstructed chromosome in those locations. So, for example, if we know that a person’s mother has an A on one chromosome, but we don’t have any information about the other chromosome, we print the SNP as being A/A. This makes the kit have “runs of homozygosity”—long regions where the person has only “homozygous” SNPs. (Homozygous SNPs are those where both of the DNA bases are the same such as C/C, and heterozygous SNPs have two bases that differ such as A/C). A drawback of this is that the parent will look inbred, but it’s not clear that a great alternative exists unless the websites that allow uploads do some engineering to allow for “half-genotyped” SNPs.
Sometime soon, we’ll allow users to select an option to print out half-missing genotypes. This will give the full information about what HAPI reconstructs, but again probably isn’t going to work for uploading to the various sites that allow uploads.
Combining different company’s raw data or different chips from the same company
Ideally all companies at all times would test people on the same set of SNPs. This would make combining data between companies or for people tested many years apart very simple. Sadly, the set of SNPs tested does vary between companies and over time for the same company. Focusing on the issue of using multiple individuals tested on different SNPs—whether from the same or different companies—a concern is that, for example, a child that is the only one that inherited, say, a red chromosome but who was not tested at a SNP that the other children were tested on can lead to a half-missing genotype in the parent. Indeed, what can happen is intermittent half-missing sites spread within a region where the parent did transmit both their chromosomes to the children. This happened in my family: my uncle was tested later than the other members of my family, has fewer SNPs than the others, and there are some places where he is the only child that inherited one of my grandma’s chromosomes. To make this more concrete, HAPI in this case may construct the following at five successive SNPs:
A/C
G/-
T/C
C/-
T/T
Interspersing “fake” full genotypes formed by repeating the one DNA base (in the above example, G at SNP 2 and C at SNP 4) to get a homozygous genotype instead of the parent’s true genotype, which may be heterozygous, can be a big problem. In general, it will lead to missed shared segments: false negatives. (If the truth is that SNP 2 is G/T and HAPI reconstructs it as G/G, that will produce a mismatch to a shared segment in a relative that does contain the T.) So, what HAPI does is, in long stretches (> 50 SNPs) of half-missing SNPs, it forms the fake runs of homozygosity, and in regions where the surrounding sites have fully reconstructed SNPs, it instead assigns what would be half-missing SNPs to be fully missing (-/-). This loss of data is less of a problem because missing genotypes—unless there are a very large number of them in a nearby location—won’t in general lead to false negative segments. Instead, the intermittent SNPs with full data will typically allow for the detection of those segments.
On the topic of using raw data from different companies, HAPI currently issues an error when a user attempts this, but there is version of the tool (linked to from the error message) that allows this. The rationale for potentially not using data from multiple companies has to do with quality control filters. Each company chooses different parameters for determining what the SNP genotypes are, and it’s possible that combining data could cause problems. However, we likely will remove this check in the future since HAPI itself detects Mendelian errors (e.g., if a parent is C/C and a child is A/A) and other forms of errors and assigns missing data to the reconstructed parent in these cases.
Other quality control filters
Related to the last section, besides detecting Mendelian and other errors, HAPI filters out SNPs where ≥ 2 individuals (the parent and/or children) are missing data. (This is increased to ≥ 3 missing SNPs when there are 6 or more children.) The reason for this is that such positions are more likely to be reconstructed as half- or fully missing and if the company determined that the SNP is missing in more than one person in a family, this is an indication that others may have erroneous data, too.
How can you use the reconstructed kit?
Several companies allow users to upload kits from other websites. This includes (alphabetically) FamilyTreeDNA, GEDmatch, Living DNA, and MyHeritage. While they may ultimately stop allowing users to upload the reconstructed data, one person wrote to say that GEDmatch accepted his HAPI reconstructed kit. Please note that this post and the availability of this tool does not indicate an endorsement of uploading these kits. Doing so may violate user agreements. However, the kits are formatted in such a way that the companies can easily prevent users from uploading them, and we hope they will not take any punitive action against users that do. (In other words, use the reconstructed kits at your own risk.)
Next steps
This version of HAPI is in “beta” (the printed kits give the current version number, 0.8b). There are several features we will add to HAPI in the near-term. One is to reconstruct the X chromosome, and, where available, Y and mitochondrial chromosomes. (SNPs on the Y and mitochondria are not provided by all companies.) Another is to produce a plot of where the parent was reconstructed on both copies and only one copy, like the one above for my grandma, and to report the percent of DNA the tool reconstructed. Also, as noted above, it will ultimately be possible to print the half-missing SNPs for positions where HAPI only reconstructed one chromosome.
Responses to questions from users
Below are a few questions people have asked.
Does Endogamy inhibit HAPI’s ability to reconstruct DNA?
The short answer is very little. The presence of one parent allows HAPI to “subtract” away that parent’s contribution to each child’s DNA. We hope to do some more analysis on this topic, but ultimately this version of HAPI (i.e., using data from one parent and multiple children) will almost certainly not attribute DNA from the parent with data to the parent being reconstructed.
What if I only have data from siblings but neither parent?
Unfortunately, unless you have, for example, ≥ 10 siblings or so, HAPI cannot to do much to help with reconstruction. The reason is that telling apart which parent is which in the data is extremely difficult. In fact, the reconstruction in large families would also have this problem, but differences in male and female recombination patterns (the subject of an upcoming blog post) allows this to work. If you’re a part of such a large family, go ahead and collect the DNA of as many of the children as you can: this tool will come!
What if I have siblings and an aunt or uncle?
Having data from an aunt or uncle would help and we can extend HAPI to work in this way. In fact, a student at Cornell is hoping to release such a tool in the near term (later this year). Once that happens, we will incorporate it onto this website, so stay tuned! (We’ll announce the release of this on our mailing list.)
What about half-siblings?
Reconstructing DNA from the shared parent of half-siblings is quite possible, and would be made even more effective if data for the non-shared parent of one or more of those half-siblings is available. While HAPI cannot yet do this—and in fact, the tool will be quite different from HAPI so will have a different name—we plan to work on this and hope to release a tool in 8-9 months. (This post is date stamped, so we’ll do our best! Again, this will be announced on the mailing list.)
What if I have a parent and two or one children?
The minimum of three children is somewhat arbitrary, and we will likely allow for the reconstruction of a parent from two children soon. Mostly, we need to do some checks to confirm that some features of HAPI work reliably in this case.
For one child, reconstruction can only provide the DNA that’s already present in that child. If someone writes with a good use case, we will likely extend to this case, but note that the reconstructed kit will be homozygous (or missing) everywhere, and may be rejected by all companies. It is not obvious how useful it will be: the matching relatives should be the same as those for the child.
Acknowledgments
This web version of HAPI extends on the original HAPI which was first written over 10 years ago. We hope to publish a paper on HAPI2 (which this website runs) later this year. For the web version, special thanks are due to Ed Williams, Debbie Kennett, and Shai Carmi who shared raw data from the various testing companies so that HAPI is able to read in data from (alphabetically) 23andMe, AncestryDNA, FamilyTreeDNA, Living DNA, and MyHeritage.
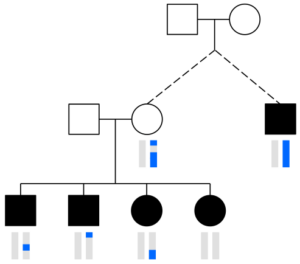
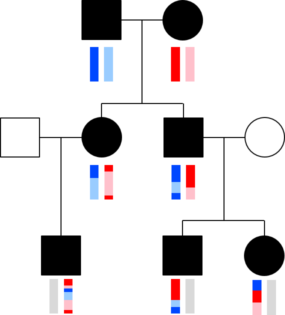
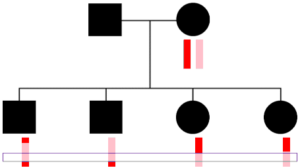
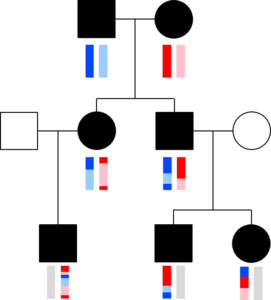 The chromosomes in germ cells are not simply an exact copy of one of the 23 chromosomes a person has, but are formed by recombination. A visualization helps capture this. The image with squares and circles shows how DNA from a couple might be transmitted to two children and three grandchildren. Here, circles represent females, squares represent males, and the vertical bars below these shapes give a colored representation of that person’s pair of chromosomes.
The chromosomes in germ cells are not simply an exact copy of one of the 23 chromosomes a person has, but are formed by recombination. A visualization helps capture this. The image with squares and circles shows how DNA from a couple might be transmitted to two children and three grandchildren. Here, circles represent females, squares represent males, and the vertical bars below these shapes give a colored representation of that person’s pair of chromosomes.