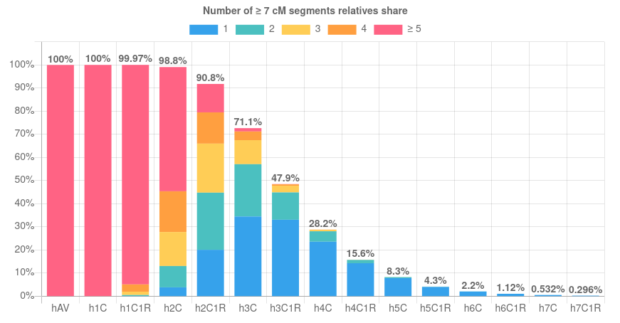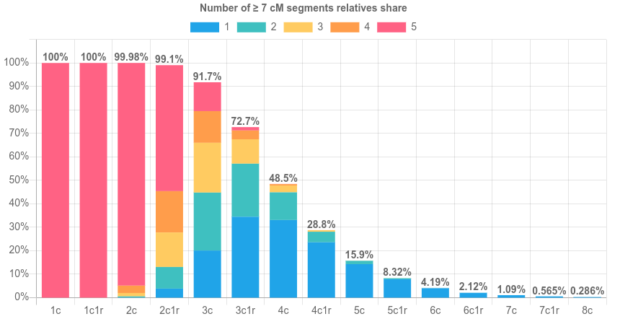
When trying to use DNA to figure out how two people are related, the closer their relationship, the easier it is. This is clear in the image below, which is from a paper that shows the rates that relatives of different degrees get classified to each of four different degrees.11st degree relatives include parent-child and full siblings, 2nd degree relatives include grandparent-grandchild, half-sibling and aunt/uncle-niece/nephew pairs. There are more types of 3rd degree and 4th degree relatives, but as an example, first cousins are 3rd degree relatives and first cousins once removed are 4th degree relatives. Genetic testing companies typically don’t try to report exact degrees of relationships for relatives because, as the image shows, it’s easy to get the answer wrong (and things get even less reliable for more distant relatives than these). However, plenty of genealogists use cM sharing numbers (and the shared cM project) to try to pinpoint how two people are related.
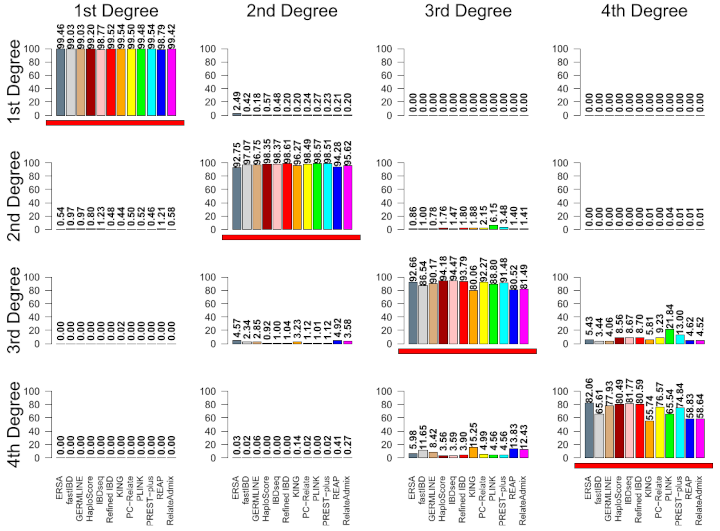
Part of a figure from a paper that calculated the accuracy of 12 relatedness classifiers. The true relationship is given on the horizontal (in each column) and the vertical (rows) gives the detected relationship from each of the 12 tools.
This post is about a new tool called DRUID for using segments that two or more siblings share with a relative to improve the determination of their relationship. Ideally we would all test our parents’ DNA (or, if possible, their parents’ DNA), since our parents are one generation closer to our non-descendant relatives than we are.2Our parents are one generation more distant than we are to our descendants: children, grandchildren, etc. This means that our parents are one degree of relatedness closer to our relatives, which makes for more reliable classification (as shown in the image above). If you do test your parents, consider testing their siblings as well since that gets you a larger portion of your grandparents’ DNA. The more siblings (including half-siblings) tested, the more of their parent’s DNA you have access to. On average two siblings inherit 75% of their parent’s DNA, three siblings inherit 87.5%, and |S| siblings inherit an average fraction of 1 – ½|S| of their parent’s DNA.
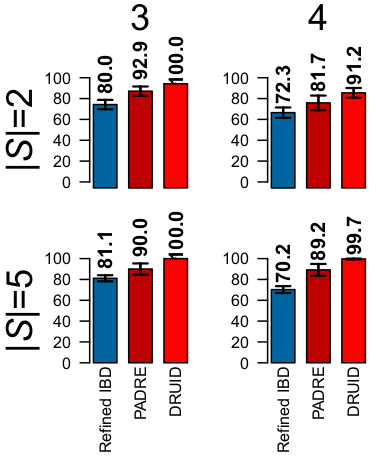
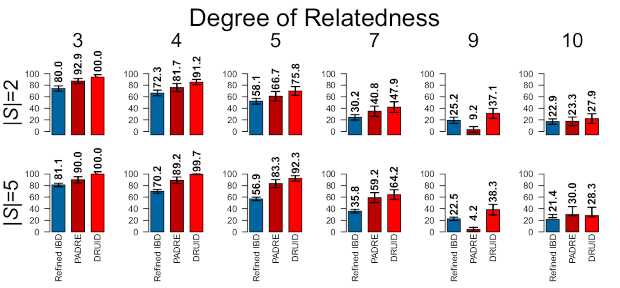
Before going into the way DRUID works, the image below shows how much DRUID can improve relationship detection.3This image is based on simulated relatives, but DRUID similarly improves real relatives. The first row of results is for two siblings, |S| = 2; the second row is for five siblings, |S| = 5; and the columns give the true degree of relationship. The bars to compare are those of Refined IBD, which analyzes each sibling independently, and DRUID (PADRE combines information but does so differently than DRUID). As an example, for 3rd degree relatives, DRUID detects 100% of the relatives correctly compared to 80% using the method that only analyzes pairs of relatives. For 5th degree relatives, DRUID detects almost 76% correctly compared to 58.1% on average when analyzing the siblings separately.
DRUID uses the same information as Refined IBD but instead of comparing each sibling to the relative independently, it combines IBD segments from the siblings to improve accuracy. This figure is from the paper that describes DRUID.
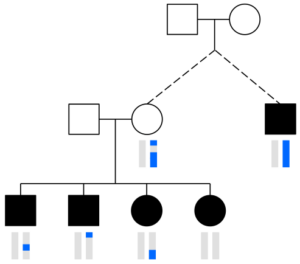
DRUID merges the segments that two or more siblings share with a relative to determine how many cMs their parent shares with that person. In this image, the distant relative (black square on the right) has one blue chromosome (blue vertical bar below) and the blue regions in the sibling’s chromosomes are segments each shares with the distant relative (siblings are the black squares and circles on the bottom left, and their chromosomes are vertical bars below each shape). DRUID assumes these segments all come from one of the parents (here, the unfilled circle above the siblings; the blue regions in the vertical bars below this circle indicate the parent’s shared segments with the relative).
Briefly, the way DRUID works is that it assumes that all segments the siblings share with the relative come from only one parent and it combines those segments together (see the image on the right). It then calculates an estimate of how much of the parent’s DNA the siblings have represented using the (1 – ½|S|) formula. DRUID assumes that this is the fraction of parent’s total cMs the siblings have inherited. So if the siblings have X cM shared with the relative, DRUID calculates T, the estimated total cM the parent shares, as T = X/F where F = 1 – ½|S|. As the image above and several other tests from the DRUID paper show, this approach improves relationship classification.
Please don’t use DRUID if your ancestry includes endogamy or if your parents are somewhat closely related to each other. In those cases, the segments the siblings share with the relative may be a mix of DNA from both parents, and DRUID will overestimate how many cMs one parent shares with the relative.
We hope you find DRUID useful and note that the effort to get this online was a collaboration with Jonny Perl who is also launching a DRUID tool.
Thanks to Monica Ramstetter who spent many hours co-developing DRUID and calculating the accuracy of academic methods for detecting relatives (a portion of which are shown in the first image above).